IPMOF
Discovering Interpenetrated Metal-Organic Frameworks

IPMOF
Discovering interpenetration in MOFs
Our publication using this library was futured on the front cover of CrsytEngComm:
Sezginel, K. B., T. Feng, and C. E. Wilmer. “Discovery of hypothetical hetero-interpenetrated MOFs with arbitrarily dissimilar topologies and unit cell shapes.” CrystEngComm 19.31 (2017): 4497-4504. DOI: 10.1039/c7ce00290d
Chemistry World article:
Algorithm deliberately entangles MOFs.
Installation
IPMOF uses Python 3.5 with required libraries listed in requirements.txt file.
You can install IPMOF by cloning the repository and running setup.py as follows:
git clone https://github.com/kbsezginel/IPMOF.git
cd IPMOF
python setup.py install
If you wish to use HPC capabilities you need to install other dependencies:
pip install -r requirements_hpc.txt
For mathutils python package (used in rotation operations), additional installation might be necessary.
In linux the following dependency for mathutils can be installed by:
sudo apt-get install python3-dev
Usage
IPMOF reads structure files from the mof folder in root directory.
The default file reader is ASE and IPMOF supports all the file formats supported by ASE. A list of the formats supported by ASE is given here.
In order to test IPMOF there is one MOF file (MOF-5 / REFCODE: SAHYIK) in the ‘mof’ directory. Using default simulation parameters homo-interpenetrated MOF-5 structure can be generated in ‘results’ directory.
Algorithm Description

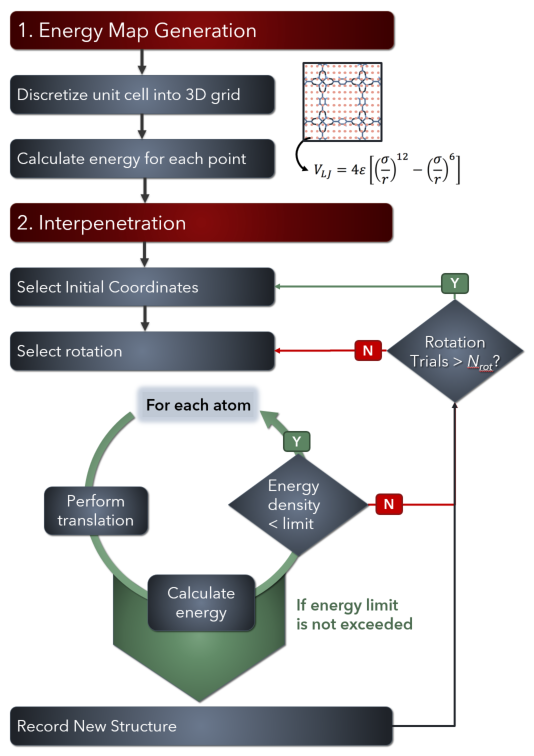
IPMOF tests whether two given MOFs can interpenetrate each other by rapidly trying different relative orientations of the two frameworks and reports the plausibly energetically favorable ones. The algorithm tries many different orientations of two given MOFs by performing rotation and translation operations according to user configurable parameters. After an orientation is chosen, its energetic favorability is calculated based on the pairwise interactions between each atom on one framework with every atom on the other framework. Overall, IPMOF can rapidly detect cases where interpenetration is impossible, and suggest ones where it may be plausible. A flowchart is provided below.
Energy Map

An energy map is a regular grid of points representing the potential energy inside the crystal unit cell, from the perspective of an atom being inserted into that space. The potential energy for each point in the grid is calculated using Lennard-Jones potential with built-in parameters provided for Universal Force Field (UFF) and DREIDING force fields.
Energy map is exported as a numpy array consisting of 3D coordinates and energy values for different types of atoms. There are four options that can be used to generate the atom list for a particular energy map:
- full: Full atom list in the force field parameters database. (103 atoms)
- uniq: Unique atoms for a given list of MOFs
- dummy: Single dummy atom with predefined force field parameters
- qnd: Simplified atom list consisting of 1 dummy atom and 10 most frequent atoms in CoRE MOF database
The runtime of the energy map generation and interpenetration tests depend on the selection of atom list in the following fashion: full > uniq > qnd > dummy. All list types except for uniq allows checking interpenetration of the passive MOF with any other MOF. The uniq energy map uses only the atoms in the given MOF set, therefore any other MOF that has other types of atoms requires new energy map. qnd and dummy enery map types were designed to work with large scale screening calculations to have one type of energy map for large amount of MOF combinations. full energy map can also be used in the same application to increase accuracy however that would result in slower runtime and bigger file sizes for energy maps.
To generate energy map type following in a command-line window:
python ipmof_energymap.py
By default this will create energy maps for each MOF file in ~/mof directory. The atom list and energy map are stored as a numpy array. This can be changed to a human readable format (yaml) by changing the energy_map_type simulation parameter to yaml.
Interpenetration
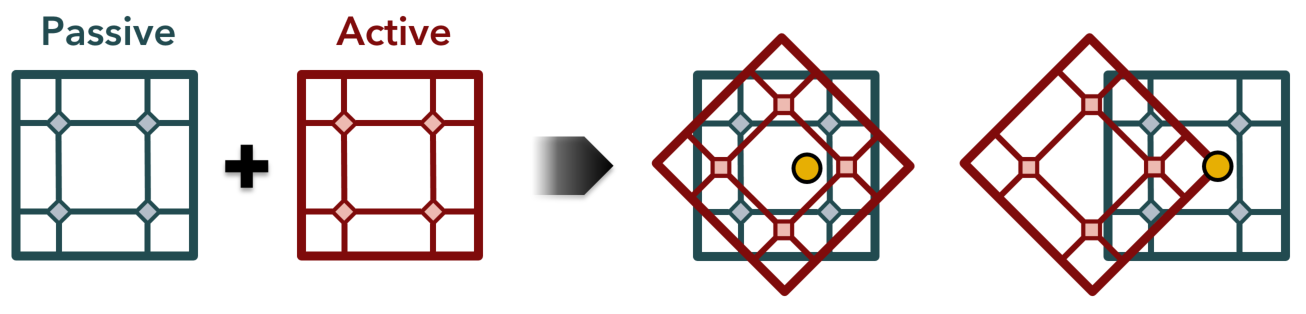
After generating the energy map for the passive MOF, an active MOF is selected to test interpenetration. The active unit cell is first aligned with the passive unit cell and then rotated around the global x, y, and z axes by increments defined by the user. Then the active unit cell is translated in x, y, and z directions, also by increments defined by the user, within the passive unit cell. After testing all orientations the most favorable ones (if any) are reported.
Energy map is required only for the passive MOF (should be in ~/energymap) and structure files (such as cif) are required for both MOFs (should be in ~/mof).
To test intepenetration type following in a command-line window:
python ipmof_interpenetration.py
Simulation summary, simulation parameters, and information on discovered structures will be exported to ~/results/*Structure1_Structure2*/results.yaml. Methods to analyze results are included in ~ipmof/analysis.py library. The structures discovered will be exported to ~/results/*Structure1_Structure2*.
Simulation Parameters
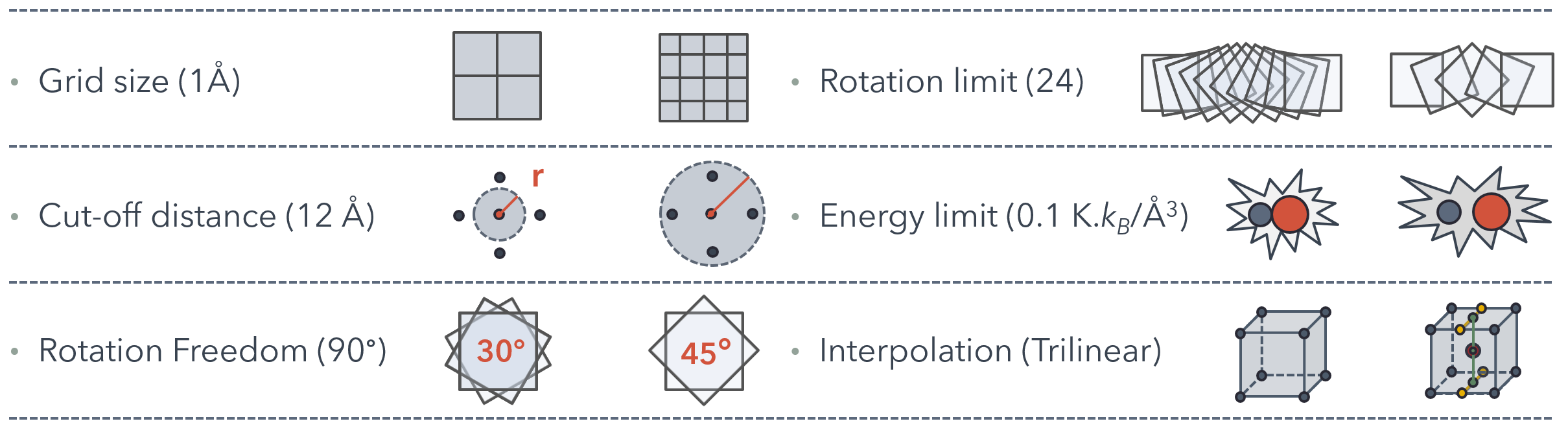
Grid size: The grid size if defined as the distance between grid points of the energy map in each dimension. The energy map is generated as a 3D rectangular grid that surrounds the unit cell. For high-throughput screening we used grid size of 1 Å for the generation of energy map.
Interpolation: When an energy value is needed at a position that does not fall exactly on a grid point in the energy map, which is the usual case, interpolation is used. For high-throughput screening we used trilinear interpolation which approximates the value of a point in a regular grid linearly using data in the lattice points.
Cut-off distance: The cut-off distance used in the calculation of interatomic potentials. Any atom pair that are further away from this distance were assumed to have no interaction. For high-throughput screening cut-off distance was taken as 12 Å
Extension cut-off distance: The extension cut-off distance is used to determine the number of extension that will be performed on the unit cell for multiple unit cell collision test. For a given distance the unit cell is extended in each dimension to make sure that it covers an area at least the size of a sphere with radius of that distance. For high-throughput screening extension cut-off was taken as 50 Å.
Rotational freedom: The increments of rotation performed on the active unit cell to obtain different orientations. For high-throughput screening only 90° rotations were considered.
Rotational limit: The number of times a rotation is performed for same initial coordinate. For high-throughput screening all possible 90° rotations (24 total) were performed for each point.
Energy limit: There are three types of energy limit defined in the algorithm: atom energy limit, structure energy limit, and energy density limit.
- Atom energy limit is the maximum allowed energy for insertion of a single atom.
- Structure energy limit is the maximum allowed energy for insertion of a collection of atoms that constitute the active structure. It is calculated by summing the insertion energies for each atom in the unit cell.
- Energy density limit is similar to structure energy limit however it is divided by the unit cell volume to make it more comparable among different structures. For unit cells with different sizes constituting different number of atoms we chose to have a universal energy limit scaled by the volume of the cell. The energy density, ρenergy, is calculated according to equation below,
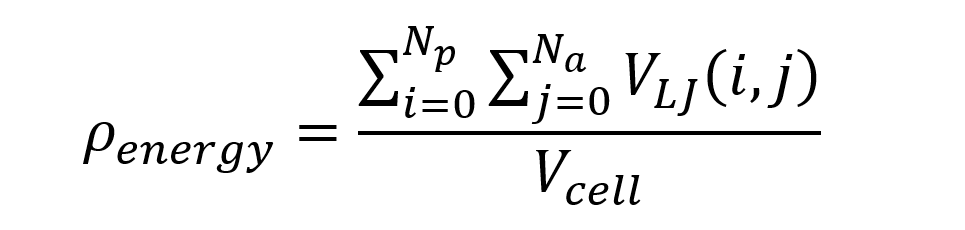
where Np and Na are number of atoms in active and passive MOFs; i and j are atom indices for passive and active MOFs; VLJ is interatomic potential (Lennard-Jones) energy, and Vcell is unit cell volume in Å3.
There are additional algorithm parameters available for user to manipulate how the structures are generated, how the data is reported etc. however these parameters do not affect the interpenetration. More information on algorithm parameters can be found in algorithm documentation.
Simulation parameters can be changed by modifying ~/settings/sim_par.sample.yaml file. The file name must be changed to sim_par.yaml after modification in order to be read by the algorithm. Without the sim_par.yaml file default simulation parameters are read from ~/ipmof/parameters.py.
Simulation directories
Default directories to read input files such as energy maps, MOF files and output result files are read from ~/ipmof/parameters.py.
If you wish to use different directories export sim_dir.yaml file into ~/settings folder with the directories you want to use. You can use methods in ~/ipmof/parameters.py to export this file.
Documents
To get a better understanding of the functions used in ipmof libraries and/or analyze your results you can go through jupyter notebook files in ~/doc/Notebooks directory.
Notebooks list:
- MOF-5 Example Run (Energymap generation and interpenetration test)
- Hetero-interpenetration Test (LEHXUT + XAMDUM02)
- Supercell Generation (LEHXUT + XAMDUM02)
- CoRE Database Usage
The ~/doc directory also contains force field parameters (UFF and DRE) and supplementary information for MOFs in CoRE database.
Candidate Structures
Candidate interpenetrated structure files can be accessed here -> IPMOF-candidates
Contact
For any questions, ideas or feedback please contact me!
Mail: kbs37@pitt.edu
Web:
Algorithm Flowsheet